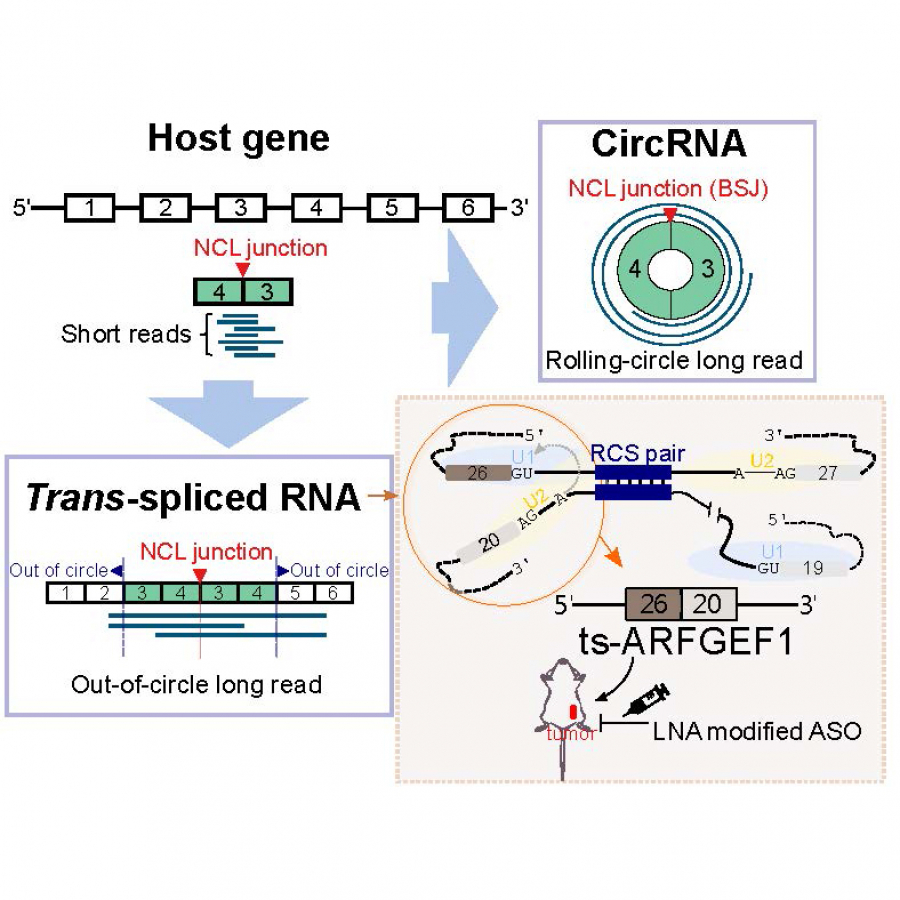
Trans-spliced RNAs (ts-RNAs) are a type of non-co-linear (NCL) transcripts that consist of exons in an order topologically inconsistent with the corresponding DNA template. Detecting ts-RNAs is often interfered by experimental artifacts, circular RNAs (circRNAs) and genetic rearrangements. Particularly, intragenic ts-RNAs, which are derived from separate precursor mRNA molecules of the same gene, are often mistaken for circRNAs through analyses of RNA-seq data. Here we developed a bioinformatics pipeline (NCLscan-hybrid), which integrated short and long RNA-seq reads to minimize false positives and proposed out-of-circle and rolling-circle long reads to distinguish between intragenic ts-RNAs and circRNAs. Combining NCLscan-hybrid screening and multiple experimental validation steps successfully confirmed that four NCL events, which were previously regarded as circRNAs in databases, originated from trans-splicing. CRISPR-based endogenous genome modification experiments further showed that flanking intronic complementary sequences can significantly contribute to ts-RNA formation, providing an efficient/specific method to deplete ts-RNAs. We also experimentally validated that one ts-RNA (ts-ARFGEF1) played an important role for p53-mediated apoptosis through affecting the PERK/eIF2a/ATF4/CHOP signaling pathway in breast cancer cells. This study thus describes both bioinformatics procedures and experimental validation steps for rigorous characterization of ts-RNAs, expanding future studies for identification, biogenesis, and function of these important but understudied transcripts.
Language
週四, 27 七月 2023 16:12