Professor Trees-Juen Chuang from Genomics Research Center of Academia Sinica has brought up an in-triguing observation regarding the risk factors of genetic mutations that may yield to diseases. This is the first time to explore the association between RNA editing activities and the damaging effect of genetic mutations at a population scale. His team brought up solid proofs using Bioinformatic Analysis and says, the “RNA editing” factor should definitely be counted when evaluating disease prone gene mutations. The study was published online in Genome Research recently.
Ever since The Human Genome Project has completed the decoding of the 3 billion DNA codes in the human genome, a brand new door was opened in the therapeutic investigations world. If we envision the human genome as a human genetic dictionary, than, scientists can use it to check for wrong spellings related biological meanings, and all sorts of genetic related questions. Thanks to the recent development in high-throughput sequencing technologies and tools, personalized genome sequencing appears much easier to obtain, and Precision Medicine seems more than realistic. The new challenging is how to deal with (or systematically analyze) various types of “Big” data, interpret the analysis results, and apply the results to search for meaningful biological answers above and beyond.
An easy way to look at the the whole DNA sequence is that there are four letters: A, G, C, T in this mighty giant dictionary. All the genes are encoded from the DNA templates based on the central dogma of molecular biology: the (DNA → RNA and RNA → protein) process. Proteins then get to perform all sorts of biological activities. However, there is a catch! If one gene is spelled wrong, the protein made accordingly may be totally off track, and sometimes, fatally wrong.
“Point Mutation” is one type of mutations that alter one letter only. For example, a position in the gene that is supposed to be “A”, somehow was turned to “G”. If a point mutation can change an amino acid, it is called as a “Missense mutation”. Missense mutations are generally categorized as risk prone. Some missense mutations are often used in our daily physical examinations.
The question is, although data tells us a certain missense mutation is related to a certain disease, it is not necessary that all those with that missense mutation would have such disease. According to the general rule of Natural Selection, shouldn’t the ones carry such a mutated gene be considered non-fit and be long gone?
Professor Chuang was puzzled and targeted the so-called “A-to-G RNA Editing” to try to resolve the mystery. It is known that A-to-G RNA Editing is triggered by a protein called ADAR. ADAR is known as an essential gene in multi-cell animals. ADAR can perform one task at the RNA level, namely, swapping an “A” to a “G”. Several studies have shown the high relationship between A-to-G RNA editing and neuronal diseases, such as autism, Amyotrophic lateral sclerosis, epilepsy, and Alzheimer's disease. Chuang’s team has studied RNA editing for many years. They had developed a tool called ICARES to detect A-to-G RNA editing and comprehensively analyzed A-to-G RNA editing across 20 multi-cell animals. (See: https://academic.oup.com/gbe/article/10/2/521/4774976)
They raised a bold assumption, maybe, A-to-G RNA editing is associated with the persistence of deleterious A/G genomic variants in a population. A-to-G RNA editing events may compensate for G-to-A genomic mutations to a certain extent and thus partially reduce the deleteriousness of such mutations, resulting in that the damaging mutations are preserved in a population and not eliminated by negative natural selection.
They collected the DNA and RNA sequencing data from 447 people and detected the A-to-G RNA editing events using ICARES. They then probed the associations between A-to-G RNA editing activities, deleteriousness of A/G mutations, and allele frequency of the mutations in a population and asked whether the observed patterns were associated with the importance of the genes/loci where the RNA editing sites were located.
As a result, for an A/G genomic variant, if G is a minor allele within a population (which implies that G is damaging), then the A allele should be edited less to prevent the conversion of A into G at the RNA level. In contrast, if A is a minor allele (which implies that A is damaging), then editing of the A allele should be promoted to compensate for the deleterious G-to-A change to a certain extent. Such a trend is stronger in functionally more important loci/genes than in less important ones.
They further extracted rare missense mutations from the 1000 Genomes Project (including more than 2,500 individuals) and asked whether RNA editing is associated with the distribution of missense variants (or nonsynonymous SNPs) in a population, especially when the missense changes are damaging. They observed that G-to-A missense mutations were much more prevalent than A-to-G ones (and other types of changes) at functionally important sites (Fig. A), in which the differences in the mutational burden of missense changes was significantly positively correlated with the deleteriousness of the changes. Moreover, the deleteriousness of G-to-A changes is significantly positively correlated with the percentage of binding motif of RNA editing enzymes at the variants (Fig. B), also supporting the association between A-to-G RNA editing and the increased burden of G-to-A missense mutations.
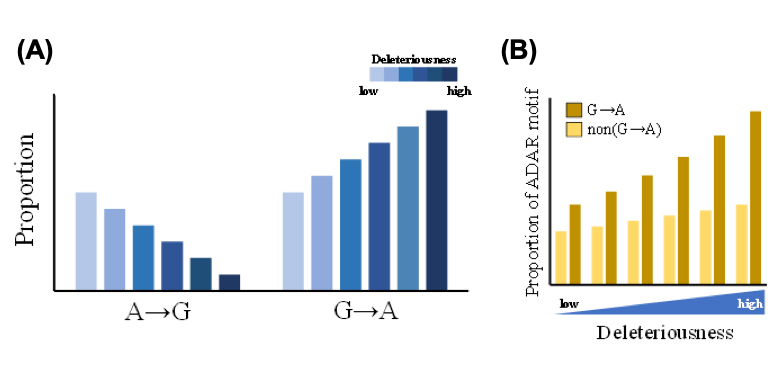 |
| (A) Correlation between the distributions of different types of rare missense mutations from the 1000 Genome Project and deleteriousness of the corre-sponding genomic changes. (B) Comparisons of the proportions of SNP sites with the ADAR motif for different deleterious effects of G-to-A rare missense mutations. |
To explain it logically, for an existing A/G missense variant, if the G-to-A genomic change has severely deleterious effects on protein function, RNA editing at this site with a higher editing level is more likely to neutralize the deleterious effect of the genomic change at the RNA level, making the deleterious effect weaker than expected. RNA editing may facilitate the escape of this existing deleterious A/G variant from negative natural selection and therefore aid the tolerance for this deleterious variant (i.e., the deleterious allele A) in a population.
Prior investigation regarding diseases and DNA mutations never considered the possibility of any down steam RNA editing effects. This study therefore provides a new insight into the role of RNA editing in the pathogenomics studies.
This study is a result of highly logical analysis and big data manipulation with bioinformatics (Fig. C). For Prof. Chuang’s group, half of the team are with pure mathematical science discipline and the other half with life science trainings. Together, they have pondered solutions to critical biological issues using data science. Prof. Chuang encourages anyone with enthusiasm and the similar backgrounds to join them.
 |
| (C) The radar chart representing the importance of the study on each of the six domains of knowledge. |
The first author of the study is Dr. Te-Lun Mai. The detailed research can be read online at: https://genome.cshlp.org/content/early/2019/09/12/gr.246033.118.full.pdf+html