Aminoglycoside antibiotics are essential drugs used in the clinical treatment of bacterial infections. These antibiotics work by binding to bacterial ribosomes, thereby inhibiting protein production. The amine group in aminoglycoside antibiotics is a critical functional group for recognizing and binding to ribosomes.
However, pathogenic bacteria may develop resistance to aminoglycoside antibiotics through given drug-resistance genes that can acetylate amine groups. As Darwin once said, "It is not the strongest of the species that survives, nor the most intelligent, but the one most responsive to change."
In evolutionary competition, some bacteria have developed aminoglycoside antibiotics to attack competitors, and some use acetylation to inactivate these antibiotics. Competitors, in turn, evolve new types of amine modifications, such as formimidoylation or iminoacetylation, to respond. Upgraded antibiotics are no longer susceptible to inactivation by acetylation and maintain a degree of affinity to ribosomes.
To explore the biosynthetic pathway of streptothricin derivatives, Professor Tsung-Lin Li from the Genome Research Center and Professor Yoshimistu Hamano from Fukui Prefectural University formed an international research team. They discovered a biochemical function of the formimidoylation or iminoacetylation enzyme (Orf1), which can produce formimidoylation or iminoacetylation of aminoglycoside antibiotics using several different substrates. In vivo gene knockout and in vitro enzyme reaction experiments confirmed this finding (Fig. 1).
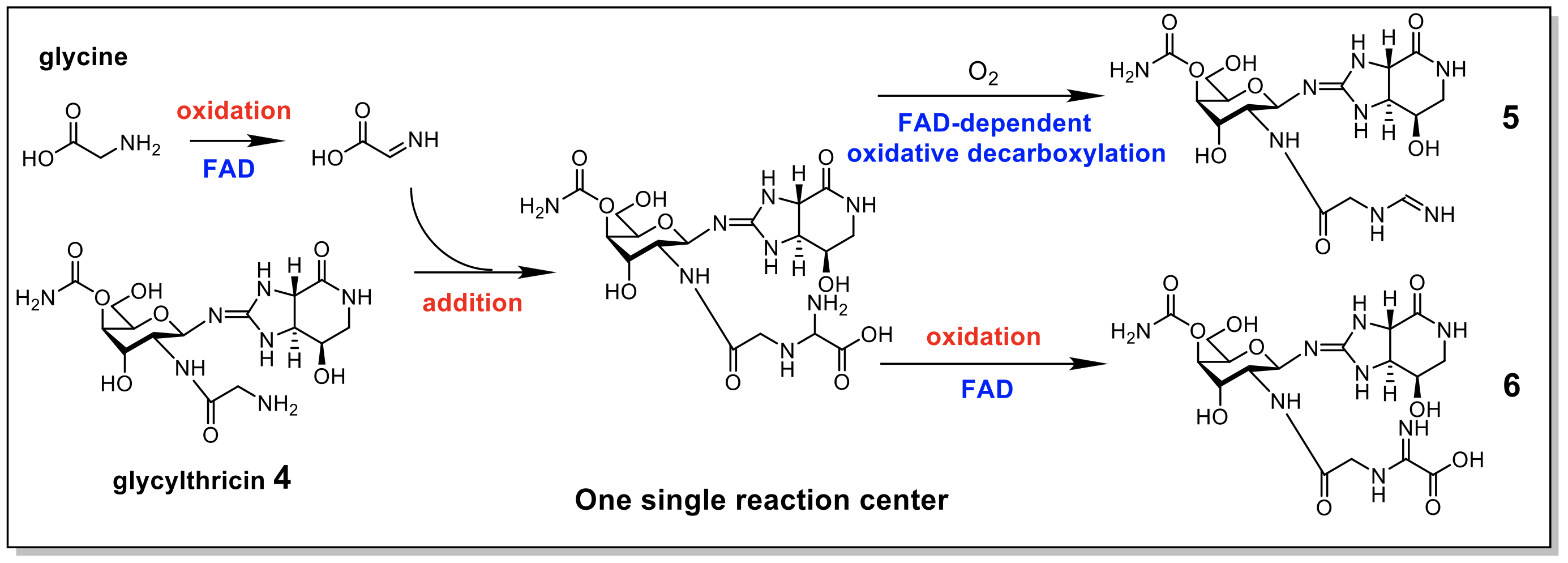 |
| Figure 1. The N-formimidoylation or N-iminoacetatylation was presumed to follow multiple steps of reactions in one single reaction site as does a typical FAD-dependent enzyme. |
To understand the mechanism of action of the enzyme, they analyzed the complex structures and found that the binding site between glycine and the streptothricin precursor is far away, making it unsuitable for addition reaction. Moreover, the intermediate product after addition is unlikely processed by FAD-dependent decarboxylation and secondary oxidation reactions (Fig. 2-A). Unexpectedly, they discovered that oxidized glycine forms the first "nitrogen-oxygen-sulfur" covalent bond bridge between the substrate and the enzyme through the sulfur atom of the enzyme cysteine (Fig. 2-B).
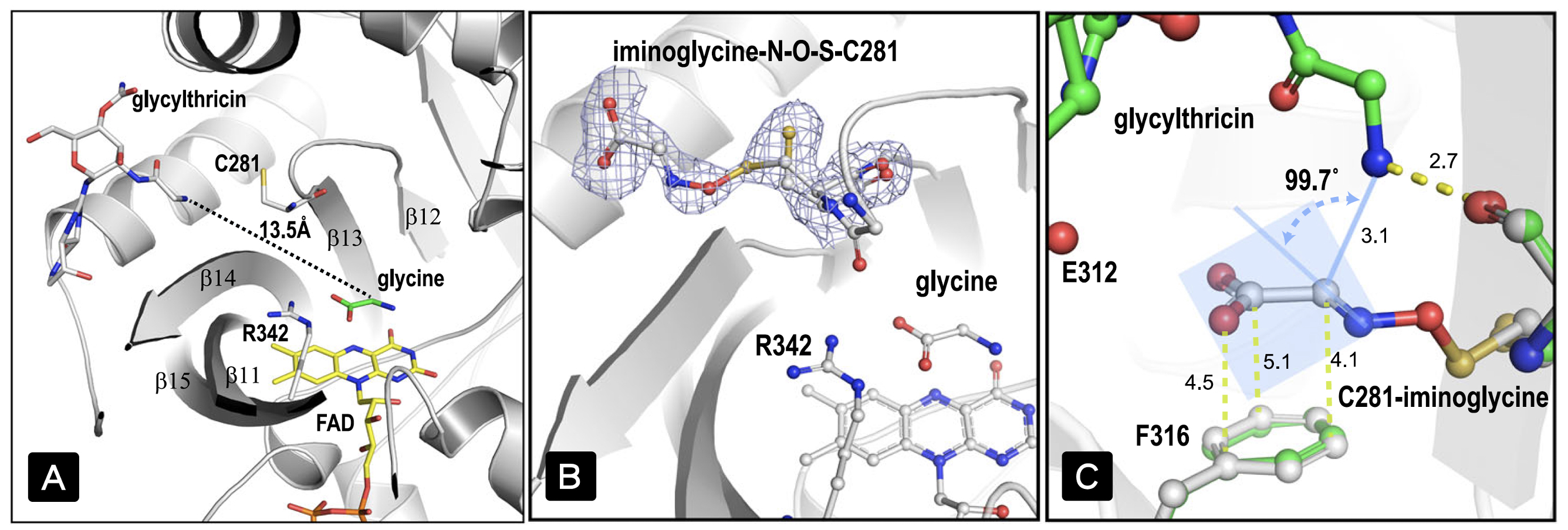 |
| Figure 2. Two substrate-binding sites sit 13.5 Å apart; the electron density region suggests a glycine imine adduct in a covalent linkage to C281 through a N–O–S bridge; the arrangement between the amine group of glycylthricin and α-carbon of the iminoacetate adduct poses a trajectory appropriate for addition reaction. |
This NOS bridge is different from the recently reported in proteins, which is a reversible post-translational modification that spontaneously forms between reactive oxygen species and lysine/cysteine and usually related to the regulation of protein function. The ligand-protein NOS bridge is the active substance formed by the oxidation of glycine via FAD, which is transported to the streptothricin precursor binding site through the intramolecular channel of the enzyme, and oxidized cysteine (sulfenic acid) residues formation of NOS bridge. There is a suitable distance and angle between nitrogen and carbon atoms in the overlapping antibiotic substrates and NOS bridge crystal structures (Fig. 2-C), which is conducive to the formation of products in the addition reaction. This is the first time an NOS bridge chemistry has been directly involved in an enzymatic reaction.
They also discovered that two amino acid residues located near the NOS bridge are crucial for generating different products based on the point mutation analysis. The mutant enzymes primary product shifted from formimidoylated to iminoacetylated antibiotics. These two amino acids may promote the elimination reaction caused by decarboxylation, resulting in bond breakage between nitrogen and oxygen. However, protein covalent bond breakage between oxygen and sulfur can regenerate oxygen-18 sulfenic acid residues by reacting different streptothricins in oxygen-18 isotope water experiment. Furthermore, after dehydration, the product formed iminoacetylation, indicating that controlling the position of bond breaking (between N-O and O-S) can dictate the enzyme to produce two different products (Fig. 3).
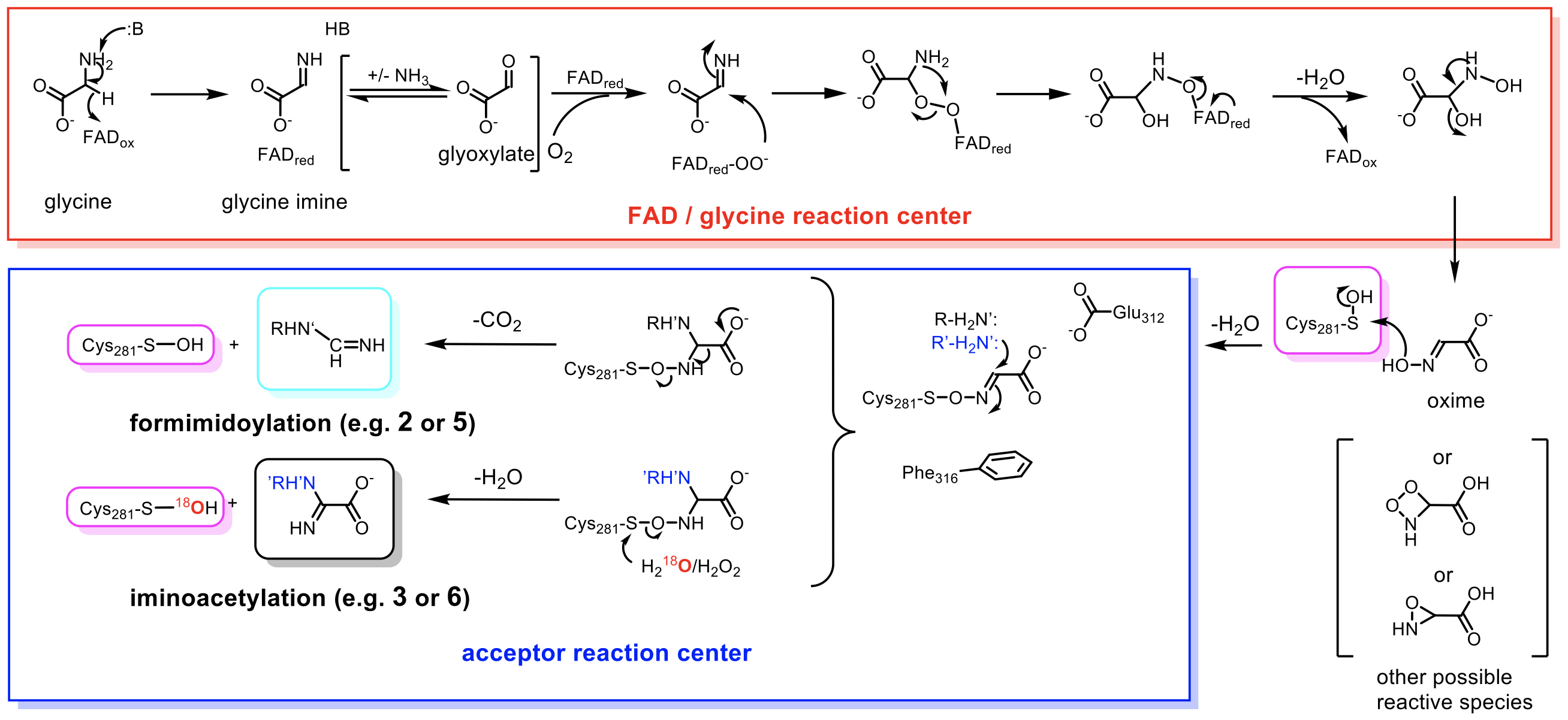 |
| Figure 3. The proposed catalytic mechanism of Orf1 based on intrinsic experimental findings. |
This is significantly different from traditional FAD-dependent enzymes through oxidative decarboxylation. Finally, they confirmed that these modifications can prevent antibiotics from being inactivated by acetylation via resistance genes. This discovery provides a new type of modification option for the development of the next generation of aminoglycoside antibiotics.
The research was published in Nature Communications, in which Dr. Yung-Lin Wang is the first author, and was completed by an international team led by Professor Tsung-Lin Li, Dr. Chi-Fon Chang from the Genome Research Center, Professor Yoshimitsu Hamano from Fukui Prefectural University, Professor Tohru Dairi from Hokkaido University, and Professor Chin-Yuan Chang from Yang Ming Chiao Tung University.
A full study“N-Formimidoylation/-iminoacetylation modification in aminoglycosides requires FAD-dependent and ligand-protein NOS bridge dual chemistry”can be read online at: https://www.nature.com/articles/s41467-023-38218-w